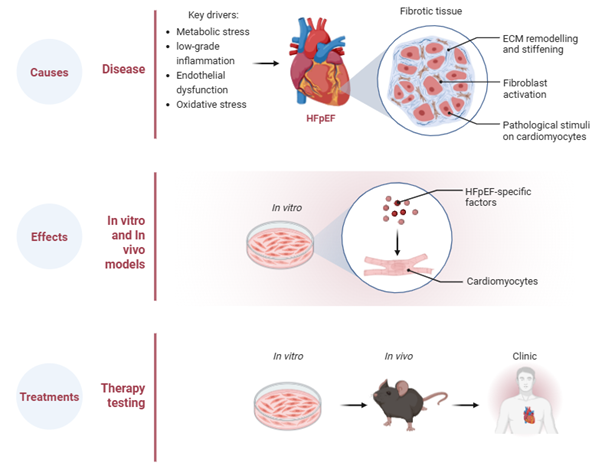
Heart failure with preserved ejection fraction (HFpEF) is characterised by diffuse myocardial fibrosis that drives ventricular stiffening, impaired relaxation and reduced exercise capacity. Despite its central role in disease progression, the mechanisms underlying fibrosis in HFpEF remain incompletely understood, and no therapies directly target the cardiac fibroblast. Importantly, the drivers of fibrosis in HFpEF likely differ from those in heart failure with reduced ejection fraction (HFrEF), where scarring typically follows ischaemic injury.
In HFpEF, fibrosis develops in the absence of large-scale myocyte loss and instead reflects chronic cardiometabolic stress, vascular dysfunction and low-grade inflammation. Increasing evidence indicates that cardiac fibroblasts undergo specific changes in HFpEF, promoting extracellular matrix accumulation and adverse structural remodelling.
Our research program focuses on defining the causes, consequences and therapeutic reversibility of cardiac fibrosis using established preclinical HFpEF models. We employ fibroblast-lineage approaches and targeted genetic tools to better understand how systemic metabolic and inflammatory stress reshape the cardiac fibroblasts.
We also investigate the functional effects of fibrosis on cardiac function and cardiomyocyte biology. Excess matrix deposition increases ventricular stiffness, disrupts normal myocardial architecture and may contribute to arrhythmias. By integrating structural and physiological assessment in experimental models, we aim to define how fibroblast activation translates into impaired cardiac function.
Importantly, our work extends beyond mechanistic insight to therapeutic testing. We are evaluating emerging antifibrotic strategies being developed globally while also advancing novel in-house therapeutic approaches designed to modulate maladaptive fibroblast signalling in HFpEF. In preclinical models, selective targeting of fibroblast-driven pathways reduces matrix deposition and improves measures of diastolic function, supporting the concept that fibrosis in HFpEF is a dynamic and potentially reversible process.
Targeting cardiac fibrosis in HFpEF